


- 收藏
- 加入书签

LC-MS/MS法测定解酒类食品中15种非法添加药物
 打开文本图片集
打开文本图片集
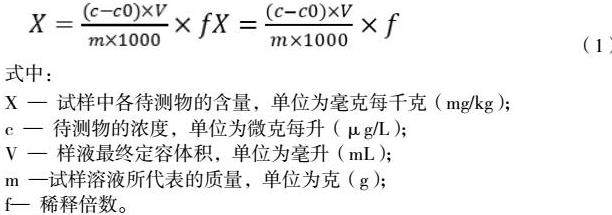
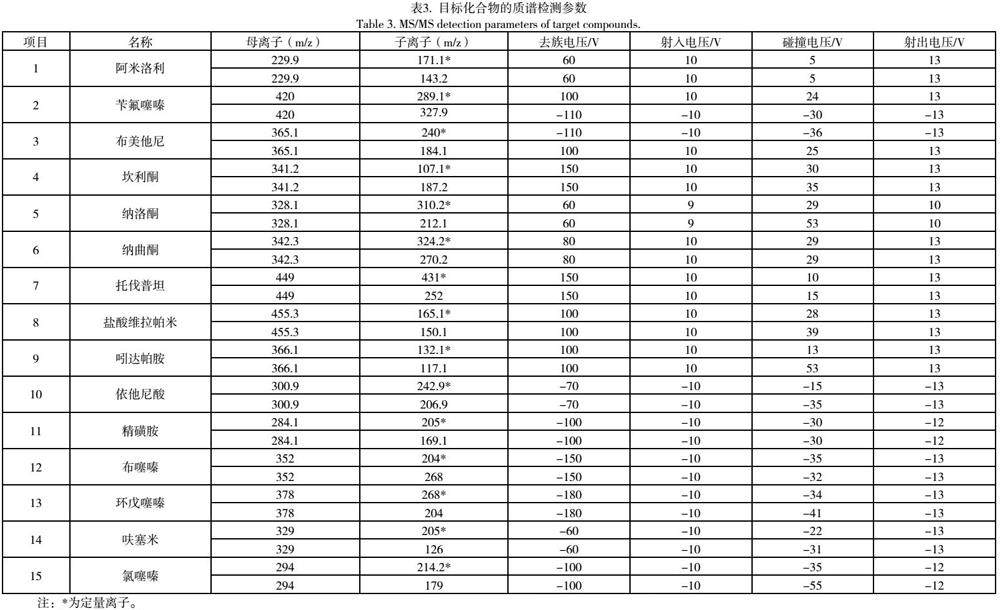

摘要 目的:建立LC-MS/MS法同时测定解酒类食品中可能添加的15种药物含量的检测方法。方法:样品中目标物分别经pH为4.5和pH为10的无水乙醇超声提取,采用Waters ACQUITY UPLC®HSS T3(2.1 mm × 100 mm × 1.8 μm)色谱柱进行分离,流动相为5mmol/L乙酸铵含0.1%(v/v)甲酸水-乙腈,梯度洗脱;流速0.4 mL/min;柱温40℃;多反应监测模式(multiple reaction monitoring,MRM)定量分析。结果:在最优条件下,该方法中所有药物成分均在10~1000 ng/mL范围内线性良好,相关系数R2均大于0.995,方法检出限均低于5 ng/mL,定量限分别低于10.0ng/mL,回收率均在80%~110%之间,连续六次平行实验结果之间的相对标准偏差均<10%。结论:该方法灵敏度高,选择性好,结果准确可靠,可同时快速筛查检测各种食品中15种可能添加的解酒醒酒类药物。
关键词:解酒食品 快速检测,LC-MS/MS
ABSTRACT: Objective: To establish a detection method for the simultaneous determination of 15 drugs that may be added to anti-alcoholic foods using LC-MS/MS methods. Methods: The target substances in the sample were ultrasonically extracted with absolute ethanol at pH 4.5 and pH 10, and separated with a Waters ACQUITY UPLC®HSS T3 (2.1 mm × 100 mm × 1.8 μm) chromatographic column. The mobile phase was 5mmol/L. Ammonium acetate contains 0.1% (v/v) formic acid water-acetonitrile, gradient elution; flow rate 0.4 mL/min; column temperature 40°C; multiple reaction monitoring (MRM) quantitative analysis. Results: Under optimal conditions, all pharmaceutical ingredients in this method showed good linearity in the range of 10~1000 ng/mL, the correlation coefficients R2 were all greater than 0.995, the detection limits of the method were all lower than 5 ng/mL, and the quantitation limit was 10.0ng/mL, the recoveries were all between 80% and 110%, and the relative standard deviations between the results of six consecutive parallel experiments were all <10%. Conclusion: This method has high sensitivity, good selectivity, accurate and reliable results, and is able to quickly screen and detect 15 possible anti-alcoholic and sobering drugs in various foods at the same time.
Keywords: Antialcoholic drugs, rapid detection, LC-MS/MS
本研究利用超高效液相色谱串联质谱仪对宣传解酒、醒酒的食品中可能添加的15种药物进行检测。喝酒伤身,长期过量饮酒会对人体健康造成损害。造成宿醉的主要原因是部分酒精会进入脑部,导致脑神经产生错觉和紊乱,为了应酬,又为了保护身体,从而各种号称醒酒解酒的食品出现了,在这些食品中,不少食品存在添加药物的嫌疑,其安全性、危害性以及用量均未经过验证,根据食品安全法,食品中禁止添加药物,在食品中添加药物是一种违法行为。
到目前为止,还没有解酒或者预防醉酒的特效药物,通常,号称能解酒的食品一般通过两种药物来达到解救的目的:一种是添加促进大脑清醒的药物,如纳洛酮、龙脑等,第二种添加利尿剂,促进酒精快速排出。目前并无食品中解酒类药物的检测方法,本论文建立了了一种宣称解酒类食品中非法添加剂的扫描检测方法。
1.材料与方法
1.1 仪器与试剂
AB 4500超高效液相色谱-质谱/质谱联用仪(配有电喷雾离子源(ESI)、Analyst采集系统及MultiQuant数据处理系统)(美国SCIEX公司),超声波清洗器(天津奥特赛恩斯仪器有限公司),XS205DU电子分析天平(0.001mg,瑞士梅特勒),ME204E电子分析天平(0.01mg,瑞士梅特勒),Milli-Q Advantage超纯水机(美国密理博),高速离心机(蜀科)
阿米洛利(98%,源叶生物),苄氟噻嗪(99.9μg/mL,广电计量),布美他尼(99.7%,北京振翔科技),呋塞米(98%,常春藤),坎利酮(98.06%,源叶生物),氯噻嗪(99.4%,广州硕普),盐酸纳洛酮(96%,TRC),盐酸纳曲酮(100μg/mL,广电计量),托伐普坦(100μg/mL,广电计量),盐酸维拉帕米(100.4μg/mL,广电计量),依他尼酸(99.9μg/mL,广电计量),吲达帕胺(97.7%,中国食品药品鉴定研究所),精磺胺(99%,广电计量检测集团有限公司),布噻嗪(100.5μg/mL,广电计量),环戊噻嗪(99.6%,广电计量),甲醇(HPLC级,上海安谱CNW),乙醇(HPLC级,上海安谱CNW),甲酸(LCMS级,fisher),乙酸铵(LCMS级,fisher),水为自制一级水(密里博),中性氧化铝(ALN)(层析级,上海国药),C18粉末胶(层析级,深圳逗点生物),乙二胺-N-丙基硅烷(PSA)(层析级,深圳逗点生物),硫酸镁(层析级,上海国药),乙酸(优级纯,广州化学试剂厂),氨水(分析纯,广州化学试剂厂)
1. 2实验方法
1.2.1溶液配制
提取液A:取无水乙醇1000mL,用浓氨水调节pH为10.2
提取液B:取无水乙醇1000mL,用磷酸调节pH为4.5
2 5mmol/L乙酸铵溶液(含0.1%甲酸):称取0.385g乙酸铵,加入1mL甲酸,用水定容至1L。
1.2.2 标准储备液的配制
分别称取阿米洛利、托伐普坦、吲达帕胺、精磺胺、苄氟噻嗪、布美他尼、呋塞米、坎利酮、氯噻嗪、纳洛酮、纳曲酮、盐酸维拉帕米、布噻嗪、环戊噻嗪各10mg置100mL容量瓶中,用乙醇水(1+1)定容至刻度,从中吸取1mL至100mL容量瓶,用乙醇水(1+1)定容至刻度,摇匀作为储备液,浓度约为1000ng/mL。
混合标准系列工作液的配制:将标准储备液分别用乙醇水(1+1)稀释至浓度为10、50、200、500、1000ng/mL,做为标准系列工作液。
1.3 样品前处理
1.3.1样品提取:
提取方式A:(用于阿米洛利、托伐普坦、吲达帕胺、精磺胺):取无水乙醇,用浓氨水调节pH至10.2作为提取液,固态试样或半固态试验充分混匀,研细;液态试验充分摇匀,称取1g(精确至0.01 g)置于10 mL容量瓶中,加提取液适量,超声提取15 min,放冷至室温,用乙醇定容至刻度,转移离心管中,20000 r/min离心3min。
提取方式B(适用于苄氟噻嗪、布美他尼、依他尼酸、呋塞米、坎利酮、氯噻嗪、纳洛酮、纳曲酮、盐酸维拉帕米、布噻嗪、环戊噻嗪):取无水乙醇,用乙酸调节pH至4.5作为提取液,固态试样或半固态试验充分混匀,研细;液态试验充分摇匀,称取1g(精确至0.01 g)置于10 mL容量瓶中,加提取液适量,超声提取15 min,放冷至室温,用乙醇定容至刻度,转移离心管中,20000 r/min离心3min。
1.3.2 样品净化
取离心后的上清液1.5mL于装有50mgALN、50mgC18、30mgPSA和100mgMgSO4填料的离心管中,涡旋混匀1min,静置5min,取上清液过0.22μm供超高效液相色谱-串联质谱联用仪测定。
1.4液相色谱-串联质谱条件
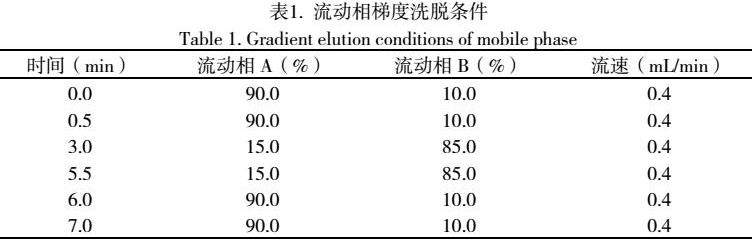
1.4.1 液相色谱条件 采用Waters ACQUITY UPLC®HSS T3色谱柱(2.1 mm × 100 mm × 1.8 μm);进样量5 μL;流速:0.4 mL/min;柱温40℃;梯度洗脱:流动相A为5mmol/L乙酸铵含0.1%(v/v)甲酸水,流动相B为乙腈,洗脱程序见表1。
1.4.2 质谱条件 气帘气流速:30 L/min;雾化气流速(GS1):50 L/min;辅助加热气流速(GS2):50 L/min;碰撞气(CAD):中等强度(medium);辅助加热气温度:500 ℃;喷雾电压:5000 V(ESI+)/-4500 V(ESI-);扫描模式:多反应监测模式(MRM)。
1.5 定性测定
a.采用优化后的仪器方法及质谱参数对样品进行扫描,如目标物无响应则样品未检出,无需定量。
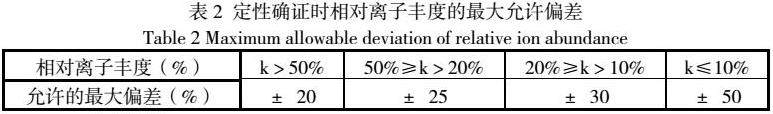
b.如扫描后出现目标物阳性则按照超高效液相色谱-三重四极杆串联质谱条件测定试样和标准工作溶液,记录试样和标准溶液中各化合物的色谱保留时间,当试样中检出与某标准品色谱峰保留时间一致的色谱峰(变化范围在±2.5%之内),并且试样色谱图中所选择的监测离子对的相对丰度比与相当浓度标准溶液的离子相对丰度比(k)的偏差不超过表2规定的范围,可以确定试样中检出相应化合物。
1.6 定量测定
将标准工作溶液按1.4所述仪器参考条件进行测定,得到相应的标准溶液的色谱峰面积。以标准工作溶液的浓度为横坐标,以定量离子的色谱峰的峰面积为纵坐标绘制标准曲线,外标法定量。
1.7 数据处理和结果计算
使用仪器自带的工作站MultiQuant数据处理系统处理数据,完成对样品中目标峰的定性定量处理。
如目标物检出按下列公式(1)计算出样品中目标物的含量:
式中:
X— 试样中各待测物的含量,单位为毫克每千克(mg/kg);
c— 待测物的浓度,单位为微克每升(μg/L);
V— 样液最终定容体积,单位为毫升(mL);
m—试样溶液所代表的质量,单位为克(g);
f— 稀释倍数。
计算结果应扣除空白值,保留三位有效数字。
2. 结果与分析
2.1 色谱与质谱条件优化
2.1.1 质谱参数优化 分别将各对照品配制成浓度为1000 ng/mL的对照品溶液,采用针泵流动注射连续进样的方式进行质谱全扫描检测,得到被测化合物一级质谱图,找出两者对应的母离子,再进行二级质谱扫描,得到相应的子离子信息。然后优化两者的碰撞能量及去簇电压等参数,使两者的母离子与特征碎片离子强度最佳,各标准品的定性和定量离子对以及最优质谱参数见表3。
2.2 提取方式的优化
由于可能添加的药物的性质各不一样,采取传统的盐析提取方式发现多种目标物质对酸或碱较为敏感,在弱酸性环境提取或弱碱性环境提取均会有部分目标物的响应很低,因而将15种药物分为两组,其中阿米洛利、托伐普坦、吲达帕胺、精磺胺、美他多辛在pH为10.2的碱性环境下提取,苄氟噻嗪、布美他尼、依他尼酸、呋塞米、坎利酮、龙脑、氯噻嗪、纳洛酮、纳曲酮、盐酸维拉帕米、布噻嗪、环戊噻嗪在pH为4.5的酸性环境下提取。在此条件下各药物的分离与定性定量均较为理想
2.3 样品净化方式的优化
由于目标药物多为油溶性药物,因而净化的首要任务就是去除脂肪的干扰,ALN去除油脂干扰的作用较强,使用C18来去除碳水化合物,并加强脂类的去除,同时想达到增加载样量和增加回收率的目的,采用PSA去除色素和有机酸,采用硫酸镁给样品快速脱水,因而最终选择了这四种填料的组合来对样品进行进化。对比净化填料PSA、C18、ALN、GCB单个及组合用量的净化效果,配比如下:
A、30mgALN+50mgC18+50mgPSA+100mgMgSO4
B、50mgALN+50mgC18+50mgPSA+100mgMgSO4
C、50mgALN+100mgC18+100mgPSA+100mgMgSO4
D、100mgALN+30mgC18+30mgPSA+100mgMgSO4
E、50mgALN+50mgC18+30mgPSA+100mgMgSO4
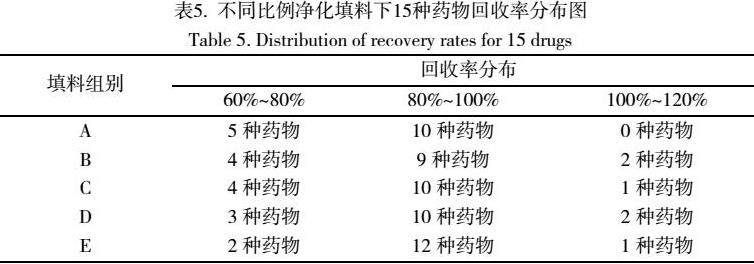
5种组合填料对宣称解酒食品中15种药物残留回收率的影响分布见表5。
通过比较,以50mgALN+50mgC18+30mgPSA+100mgMgSO4作为分散萃取净化填料的时候,15种药物中有12种药物的回收率在80%~100%之间,净化效果和回收率最佳。
2.4方法学验证
2.4.1方法的测定范围(线性范围)
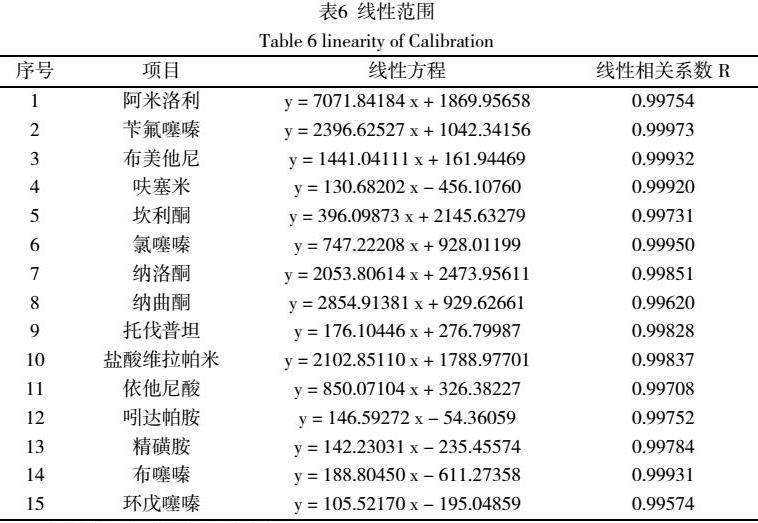
以混合标准系列工作液的浓度为横坐标,峰面积为纵坐标,绘制标准曲线,15种药物在50μg/mL ~500μg/mL范围内相关系数R2均大于0.990,方法线性关系良好,可用于食品中解酒类药物的检测
2.4.2方法的检出限和定量限
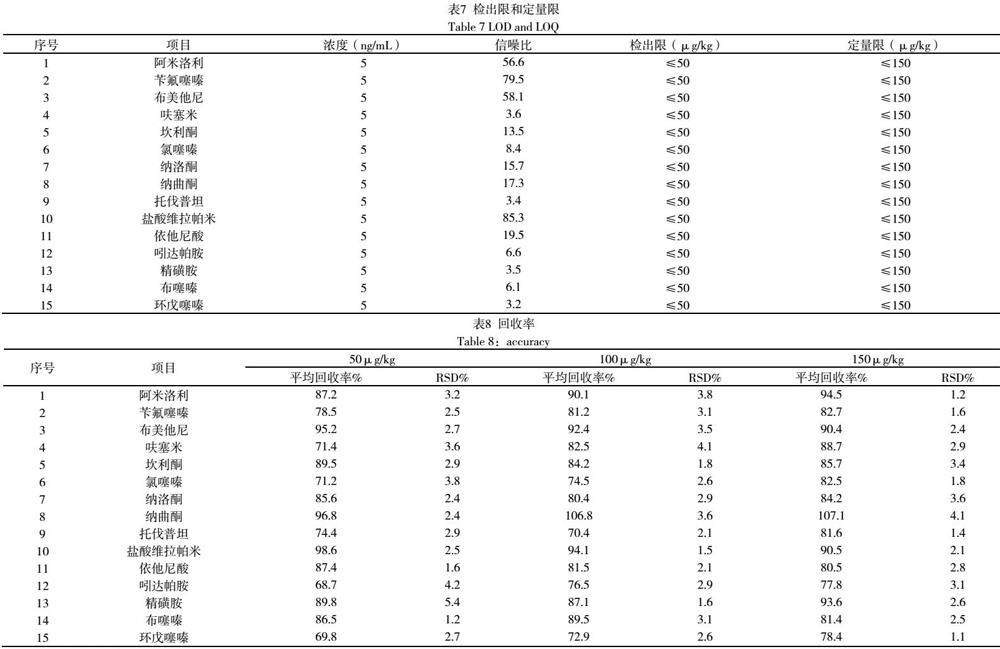
按本前处理在空白基质添加一定浓度的标液,以信噪比为3时的浓度作为检出限,信噪比为10时的浓度作为定量限,结果如表7所示。解酒类药物一般浓度在1000mg/kg以上,本方法所有药物的检出限、定量限都远低于该浓度,本方法检出限、定量限能满足预期用途,方法能用于食品中解酒类药物的检测。
2.4.3方法正确度和重现性精密度
以未检出阳性成分的某品牌桑葚解酒片为基质,分别进行50μg/kg、150μg/kg、500μg/kg 3个添加水平进行添加回收率和精密度的实验。按试样制备的方法加入适量的对照品,按照优化好前处理检测方法进行平行测定6次,计算回收率,结果见表8:从结果可知,方法所有药物的回收率均在60%~120%范围内,平行性良好,相对标准偏差均小于10%,方法准确可靠,适用于食品中解酒类药物的检测。
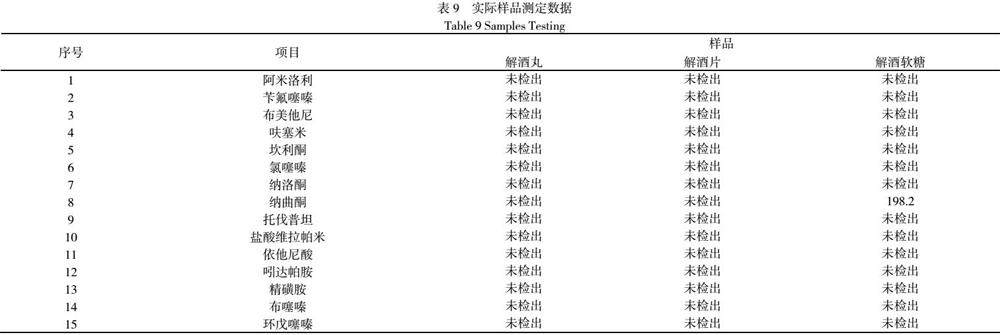
2.4.5实际样品测定
在市场随即采购了市售姜黄色解酒丸、解酒片、解酒软糖各一批,按照该方法开展检测工作,15种药物残留的检出结果如表9(单位μg/kg):
3.结论
研究建立了超高效液相色谱串联质谱法同时快速检测宣称解酒食品中可能添加的15种药物的方法。方法灵敏度高,准确可靠:方法可以在7min内完成15种药物的检测,检出限均低于50μg/kg,在50μg/mL ~500μg/mL范围内线性良好,回收率好,结果准确可靠,实际样品检测表明该方法可应用于不同基质解酒类食品中该类药物的定性、定量检测。
参考文献
[1] 刘静,吴琼,王超,液相色谱-离子阱质谱法同时测定减肥类保健食品中非法添加的12种化学药物,食品安全质量检测学报,2021.12(18)
[2] 林雨青,刘正才,郑子威,等 超高效液相色谱-串联质谱法同时测定保健食品中66种非法添加药物的含量,中国口岸科学技术,2022.4(04)
[3] 王艳红,王广珠,张任男,等 HPLC-MS/MS法同时测定止咳平喘类中成药中添加的7个化学抗菌类物质,药物分析杂志,2021,41(08)
[4] 曹美萍,朱青,徐苗,等 固相萃取-超高效液相色谱法同时测定辣椒制品中非法添加苏丹红染料,食品安全质量检测学报,2021,12(12)
[5] 邱慧,毛盛芳,UPLC-MS/MS法同时测定性保健食品中6种PDE5型非法添加物的含量,食品与药品,2019,21(03)
[6] 金舒,杨敏智,申兰慧,HPLC-DAD法同时测定降压类保健食品中非法添加的12种化学成分,药学与临床研究,2019,27(01):25-28
[7] 郭旭光,徐晓楠,邓迎春,等 液相色谱-串联质谱法同时测定清咽润喉类保健品中非法添加的沙丁胺醇和二氧丙嗪,中国卫生检验杂志,2021,31(03)
[8] 李卓,陈玉龙,孙晓,等 超高效液相色谱-串联质谱法同时测定减肥类保健食品中10种非法添加利尿类及泻下类药物,食品工业科技,2020,41(22):214-220
高效液相色谱法测定保健食品中β-烟酰胺单核苷酸,中国食品添加剂,2021.32(11)。
[9] 郭健博,薛晓文,刘开,等 超高效液相色谱-串联质谱法同时测定清咽类保健食品中14种β2-受体激动剂非法添加物,食品安全质量检测学报,2020,11(19)
[10] 曾曦,戚平,汪宁,等 HPLC-MS/MS法同时测定口服液保健食品中非法添加的降压和壮阳抗疲劳类药物,农产品加工,2020(18)
[11] 邓晶,金蔚,施祖灏,超高效液相色谱串联质谱法同时测定保健食品中的66种非法添加物,食品安全质量检测学报,2020,11(15)
*通信作者:何晓峰,高级工程师,主要研究方向为食品安全质量与检测

 京公网安备 11011302003690号
京公网安备 11011302003690号


