


- 收藏
- 加入书签

EnsPPISp:一种基于序列的蛋白质-蛋白质相互作用位点预测方法
 打开文本图片集
打开文本图片集
摘要:蛋白质在各种生物过程中扮演着重要的角色。蛋白质-蛋白质相互作用位点是蛋白质行使生物功能的重要锚点,所以蛋白质-蛋白质相互作用位点的准确预测对生物学、医学等学科都直观重要。蛋白质-蛋白质相互作用位点预测的传统生物学方法是理解生化过程机制的重要手段,但是这种方法费时费力。随着深度学习技术的发展,近年来已经出现了许多深度学习方法来预测蛋白质-蛋白质相互作用位点,但是如何有效地挖掘蛋白质的特征并增强预测地性能依然是一项挑战。本文提出了一种名为EnsPPISp的新型集成网络。该网络集成了TextCNN,Bi-LSTM和TransformerEncoder作为蛋白质生物特征提取器。同时本文提出的方法使用了预训练蛋白质语言模型ProtT5来提取蛋白质的大量潜在信息。该模型在选择的基线数据集上实现了先进的性能。
关键词:深度学习;机器学习;蛋白质序列
0引言:
虽然蛋白质在各种生物过程中发挥着关键的作用,但是它们却很少单独地执行功能。蛋白质-蛋白质相互作用(PPI)对蛋白质行使功能非常重要,它是一种蛋白质之间的物理接触。蛋白质之间结合的残基就是蛋白质-蛋白质相互作用位点(PPIS)。鉴于PPI的重要性,蛋白质-蛋白质相互作用位点预测的重要性不言而喻。
尽管现在有很多的数据库可以查询到一些蛋白质结合位点数据,但是新发现的蛋白质在不断增多,这些蛋白质结合位点的数据并没有被精确预测和收录进数据库中。目前传统的生物实验方法非常费时费力,好在过去二十年已经出现了许多计算方法来预测PPIS[1],比如利用机器学习或者深度学习技术来预测结合位点,为传统的生物实验方法提供了指导与补充。
Zeng等人开发的DeepPPISP利用滑动窗口捕获了需要预测残基及其周围残基的局部信息,并与整个蛋白质的全局信息结合通过TextCNN来预测蛋白质-蛋白质相互作用位点,其直接利用窗口长度内残基特征向量的串联来代表中心残基,这种方法忽略了来自不同相邻残基的目标残基的影响。Li等人引入注意力机制来改善了这一点,并开发了AttCNNPPISP,通过注意力机制目标残基与不同相邻残基的相关性得以建立。MLP-Mixer在相互作用位点预测方面也有应用[2]。
尽管以上方法已经在PPIS预测上解决了一些问题,显著了提升了预测的性能,但是如何有效地利用蛋白质的各方面信息、将各方面特征进行有效地组合以及如何缓解数据的不平衡性带来的负面影响依然是艰巨的挑战。本文提出了一种使用蛋白质序列信息,结合多种模型的集成学习网络EnsPPISp。
1数据集概览:
本文使用4个基准数据集来评估方法的表现。其中三个基准数据集是Dset_186,Dset_164,Dset_72,所有的这三个数据集均来自PDB数据库。本文训练和验证模型所用到的数据集来自DELPHI的工作,叫做Train9982。DELPHI从Zhang提出的数据集上进行了处理,其在保留下来的蛋白质信息上运行CD-HIT,保证没有序列的相似性高于25%,确保了数据的尽可能多样化。为构建一个适用于训练模型的高质量数据集,根据相互作用位点数的比例对序列进行筛选,选择结合位点比例较高的蛋白质序列,最后筛选得到5742 条序列。
2.特征选择:
选择合适的特征来进行建模是深度学习中的关键步骤。本方法使用蛋白质基本信息、蛋白质物化信息、进化信息、领域特征、可及表面积和ProtT5来编码蛋白质序列中的每个残基。这五部分特征的概述和选择过程如下所述:
(1)蛋白质基本信息:
本文所提到的蛋白质基本信息包括蛋白质长度和氨基酸残基相对位置。蛋白质序列中每个位置的氨基酸残基的位置信息可用一个二维向量表示。因此用这二维向量记录每个氨基酸残基的位置信息。
(2)进化信息:
PSSM是一种用于描述蛋白质序列进化保守性和预测蛋白质结构、功能的工具, 记录了在一个特定位置上每种氨基酸出现的概率或得分。其蕴含的进化信息已经在很多工作中被证明是有效的。通过 Psi-BLAST算法进行三次迭代,设定E值阈值为0.001,对uniref50数据库进行搜索。最后为每个氨基酸残基生成一个20维的特征向量。
(3)领域特征:
领域特征表示指的是指定位点的序列位置信息。通过计算该位点前后各 10个位点,共 21个位点的领域窗口,统计窗口内每种氨基酸的出现频数,以及 5 类氨基酸(非极性、极性、带电、带正电和带负电)的频数。当指定位点位于序列的起始或末尾,导致窗口大小不足 21 时,则按实际窗口大小进行计算。最终,为每个氨基酸残基构建一个 25 维的特征向量。
(4)可及表面积:
鉴于相互作用位点通常位于溶剂可及性较高的区域,本文通过式 4-2 计算蛋白质序列中每个氨基酸残基的可及表面积。其中,�代表长度为�的蛋白质序列中的第�个氨基酸残基,�表示该残基属于 20 种氨基酸中的第 �类,���(�)表示第�个氨基酸残基的相对溶剂可及性,������(�)代表第 �类氨基酸的最大可及表面积。
(5)ProtT5 Encoding
ProtT5-XL-U50是一个基于Transformer架构的预训练模型,以蛋白质序列作为输入,通过Masked Language Modeling等任务进行深度学习训练。由于该架构内嵌了自注意力机制,能够捕获序列中各个残基之间的复杂依赖关系,而位置编码技术则有效解决了自注意力机制[3]在处理有序数据时可能存在的顺序丢失问题,从而使得ProtT5能够有效地提炼出蛋白质序列地全局语义特征。本问使用ProtT5为蛋白质序列中每个氨基酸残基生成1024维的全局语义信息。在我们后续将ProtT5与其它预训练蛋白质语言模型比较的过程中发现,ProtT5更适合用来表征蛋白质-蛋白质结合事件的氨基酸序列。
3.模型设计:
本文提出用来预测蛋白质-蛋白质结合位点的模型EnsPPISp主要框架如图1所示。这是一个集成学习模型,学习了蛋白质的多方面特征。由于不同的深度学习模型有其各自的优势,可以捕获蛋白质的多方面特征,因此我们在模型的主干设计了一个由TextCNN、Bi-LSTM和Transformer Encoder三个部分组成的集成框架,用来捕获蛋白质58维生物信息的全局特征。氨基酸残基的局部信息对于PPI位点的预测十分重要,本方法使用一个窗口大小为7的滑动窗口来捕获蛋白质序列上每个残基的局部特征,为每个残基生成了一个58 * 7D的局部特征矩阵。本方法事先微调了ProtT5并用它来为每个需要预测的蛋白质生成了1024维的全局上下文特征。这些特征包含了蛋白质的大量潜在信息和相互依赖关系。
本方法为蛋白质生物信息设计了一个集成框架来捕获其全局特征。不同的深度学习模型有其自身的优点,利用这些优势的互补,模型可以相对全面地捕获到蛋白质序列的全局信息内容。
CNN最初设计用来进行图像处理, TextCNN可以被用来捕获蛋白质的全局信息。TextCNN中应用了不同大小的卷积核,以用来捕获不同数量相邻氨基酸之间的关系。最大池化层被应用用来为特征进行降维和汇聚。最大池化层的输出将被串联在一起作为蛋白质全局特征的一部分。
蛋白质序列的序列特征对于PPI位点的预测也很重要。蛋白质序列中一些残基可能会与一些遥远位置的残基有关联,因为在3D结构中,这两个氨基酸可能就非常接近,可是在1维的序列中被分开到两个相对较远的位置。Bi-LSTM是一种递归神经网络,在处理顺序相关的预测任务时可以表现出卓越的性能。Bi-LSTM可以获取序列正向和反向的信息,这十分契合蛋白质序列中氨基酸残基同时受前后残基影响的情况。本文对传统的Bi-LSTM网络稍作了修改并堆叠了三层Bi-LSTM网络,并将每层输出的前向信息和反向信息相加来增强特征。
同时该模型也使用Transformer编码器来捕获蛋白质序列的全局语义特征,全局语义特征中蕴含着丰富的特征信息,这些特征信息可以很好地填充CNN与LSTM没有捕获到的潜在缺失特征。
本方法使用长度为7的滑动窗口来捕获每个蛋白质残基附近的局部信息,这些局部信息在通过TextCNN后作为Query,全局信息同时作为Key和Value,这些信息在交叉注意力的机制下深度互联,充分融合为能较好地表征蛋白质生物信息的特征向量。这部分特征向量与预训练蛋白质大模型ProtT5产生的全局语义特征拼接,完成上游的特征提取聚合工作,在此之后特征被馈送到下游的胶囊神经网络。
主胶囊层由多个主胶囊组成,接收前一层高维的编码信息,并利用一组权重共享的滤波器提取和组合局部特征模式。每个胶囊都会输出一个向量,该向量不仅包含其对应区域的特征信息,还编码了潜在的高级属性。经过主胶囊层的八个胶囊处理后,每个氨基酸残基被编码成一个大小为 8×128 的矩阵,其中包含八个 128 维的胶囊向量,并作为分类胶囊层的输入。分类胶囊层由 16个分类胶囊构成,采用动态路由机制和非线性变换 squashing,对主胶囊层输出的胶囊向量进行更高级别的特征抽象。最终,每个氨基酸残基被编码为一个 16 维的向量,该向量经过全连接层并通过 sigmoid 激活函数,完成对相互作用点概率的预测。
4结果分析:
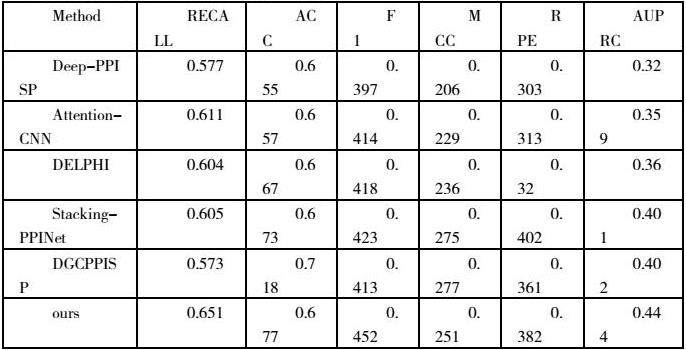
本文提出的方法相较于其他基于序列的方法,取得了优异的成绩。在Dset_186_72_PDB164数据集上,我们在F1,AUPRC这两项用于评估不平衡数据集下模型性能的指标上超过了目前基于序列的方法中最好的方法DGCPPISP,表明我们的模型在相互作用位点的识别上有优秀的性能,但是我们在另一项较为重要的指标MCC上较其他方法略有不足。
总结:
针对蛋白质-蛋白质相互作用位点预测这一问题,本文提出一种集成模型,用以研究基于序列的相互作用位点预测问题。该模型选择7种蛋白质生物特征,结合预训练模型ProtT5产生的特征。使其通过卷积神经网络提取相关的局部特征,采用双向长短期记忆网络捕获序列之间的长距离依赖关系,使用Transformer编码器为蛋白质序列进行全局建模。本方法还使用长度为7的滑动窗口捕获单个残基附近的局部信息,之后,引入注意力机制对全局信息和局部信息进行加权融合,使模型能够集中关注全局信息和局部信息之间的关联系,忽略不相关信息,从而优化预测效果。实验结果表明所提出的算法在Dset_186_72_PDB164数据集上F1分数(F1-score)和精确率-召回率曲线下面积(AUPRC)高于现有的基于序列的方法。可以看出所提出模型的优越性,为蛋白质-蛋白质相互作用位点预测提供了有效的解决方案。未来的工作中,将尝试使用不同的特征选择方法进行特征选择,采用新型的图神经网络进一步改进模型的性能。
参考文献
[1]Aumentado-Armstrong, T.T., Istrate, B. & Murgita, R.A. Algorithmic approaches to protein-protein interaction site prediction. Algorithms Mol Biol 10, 7 (2015).
[2]徐玉龙.融合MLP-Mixer的深度学习模型在蛋白质相互作用位点预测中的研究[D].云南大学,2023.
[3]吴隆鑫.基于注意力机制的DNA结合蛋白以及蛋白质作用位点预测[D].厦门大学,2021.DOI:10.27424/d.cnki.gxmdu.2021.000539.

 京公网安备 11011302003690号
京公网安备 11011302003690号


