


- 收藏
- 加入书签

基于生物信息学卵巢癌转移相关预后风险模型的构建
 打开文本图片集
打开文本图片集
摘要:目的:近年来在卵巢癌中发现了几种具有不同预后的分子亚型。然而,在卵巢癌中尚未充分研究转移相关基因分子亚型分析。方法:差异分析获取转移相关预后基因,根据表达水平对卵巢癌患者进行聚类分析,以获得聚类分型。采用LASSO-Cox回归分析建立预后风险模型,计算风险评分,并探讨预后风险模型的临床价值。结果:鉴定出58个预后基因,根据其表达水平对TCGA数据集进行聚类分析结果可得为2个分型,具有不同的预后。使用LASSO回归构建含个30基因的预后模型,对多个数据集预后具有较好预测。结果:该预后模型对卵巢癌患者的预后具有较好预测作用。
关键词:转移;卵巢癌;分子亚型;预后。
卵巢癌(oc)是妇科最普遍的恶性肿瘤之一,且死亡率最高[1]。2020年,全球约313,959例新发病例和207252例死亡病例,且发病率、死亡率被预测呈上升趋势[2]。卵巢癌早期常表现为腹部不适、恶心、腹胀,常常被误以为胃肠道疾病而被忽视[3],多数OC患者被诊断时即为晚期,且具有广泛转移。肿瘤转移与预后不良密切相关,也是卵巢癌患者死亡的主要原因。因此,探索新的生物标志物来抑制肿瘤转移至关重要。
1.材料和方法
1.1 OC 数据集和预处理
具有完整临床信息注释的开放卵巢癌基因表达数据集从公共数据库Gene Expression Omnibus(GEO)和TCGA下载。
1.2 转移相关基因的获取
在GSE178913、GSE143897数据集中使用limma包[4]进行差异基因分析,以调整P值<0.01,|logFC|>1为标准筛选差异基因,两者取交集获取相同基因,对其进行单因素Cox分析,P值取0.05,共筛选出个58个影响预后基因。
1.3 转移相关基因的无监督聚类
根据这58个基因的表达水平,使用具有最优k均值聚类的ConsensusClusterPlus R包[5]将对TCGA数据集进行聚类分析,并循环了1000次以确保分类的稳定性,最终确定了两亚型,同时进一步对GSE9891队列进行聚类分析,同样确定了两亚组,证明其聚类可重复性,并使用Kaplan-Meier方法计算不同亚组之间的总生存期(OS)分析。
1.4 功能和途径富集分析
从MSigDB数据库下载了“c2.cp.kegg.v2023.1.Hs.symbols”的基因集,以运行GSVA富集分析[6],以确定分析两亚组之间生物学功能差异的不同途径。
1.5 与转移基因亚型相关的差异表达基因
两亚组之间的差异表达基因(DEGs)采用limma R包的经验贝叶斯方法鉴定,以调整P值<0.01,|logFC|>1为标准筛选差异基因。
1.6 预后模型的建立
使用Lasso Cox回归模型[7]筛选出30个转移相关基因以确定最佳预测模型,并选择这些基因进一步计算每个患者的风险评分。
根据评分的平均值,将TCGA样本分为高低转移评分组。然后使用survminer R包对两组进行生存分析。最后,利用timeROC曲线进行分析,获取曲线下面积(AUC)值,评价特征的预测能力。计算GSE9891及GSE140082数据集的化疗评分,并用timeROC曲线评价特征的预测能力。
1.7统计学
用对数秩和卡普兰-迈耶检验绘制生存曲线。95%置信区间(CI)和风险比(HR)使用单变量Cox回归模型计算。在组间比较中,P<0.05被认为具有统计学意义。R 4.3.1软件用于数据处理。
2.结果
2.1 卵巢癌中的转移相关亚型
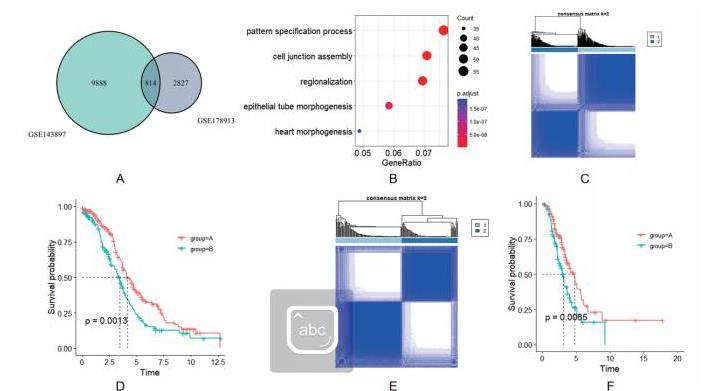
分别对GSE178913、GSE143897数据集中转移灶与原位灶使用limma包进行差异基因分析,两者取交集共获得814个共同基因(图1.A)。GO分析提示多与细胞连接、上皮形态发生、细胞功能划分相关等细胞基质相关生物活动(图1.B),TCGA卵巢癌数据集进行单因素Cox回归分析,获得了58个影响预后基因。根据58个基因表达水平对数据集进行无监督聚类分析,确定了两种亚组(A-B)(图1.C),Kaplan-Meier生存分析表明两组间存在明显差异(图1.E),A组在中位生存时间方面表现出显著优势。使用“共识聚类”R包对GSE9891进行无监督聚类分析以明确聚类稳定性,并再次明确确定了两亚组(图1.E),这两亚组的生存率同样存在显著差异(图1.F)。
转移基因与转移亚型分组。(A)两数据集差异基因韦恩图。(B)转移基因GO分析。 (C)TCGA数据集的聚类分析。(D)TCGA数据集两亚组间生存分析。(E)GSE9891数据集的聚类分析 (F)GSE9891聚类亚组间生存分析。
2.2化疗亚组生物学功能差异
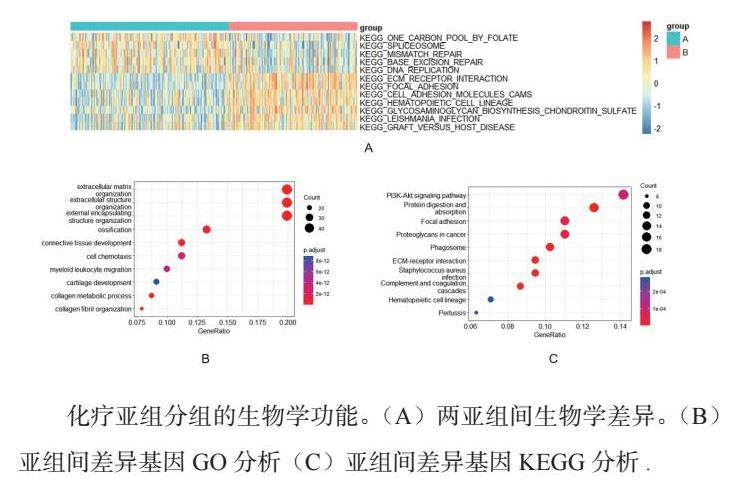
为了更好地了解两亚组之间的生物学差异。首先对亚组进行GSVA富集分析,以检查其功能和生物学差异。结果表明,A组主要与错配修复、DNA修复、碱基切除修复相关,B组主要与基质受体作用受体、 焦点粘附、细胞黏附分子。A组主要与DNA修复相关,抑制癌变,B组主要与细胞基质相关,细胞基质与肿瘤进展、侵袭、化疗抵抗密切相关,因此A组较B组具有更好的预后(图2.A)。采用limma R包鉴定两亚组间差异基因(DEGs),共获得256个基因(图3.A)。DEGs就GO分析提示细胞基质活动,KEGG分析则表明多富集于细胞基质相关通路,进一步确认聚类亚组与转移密切相关。
化疗亚组分组的生物学功能。(A)两亚组间生物学差异。(B)亚组间差异基因GO分析(C)亚组间差异基因KEGG分析.
2.3预后模型的建立与验证
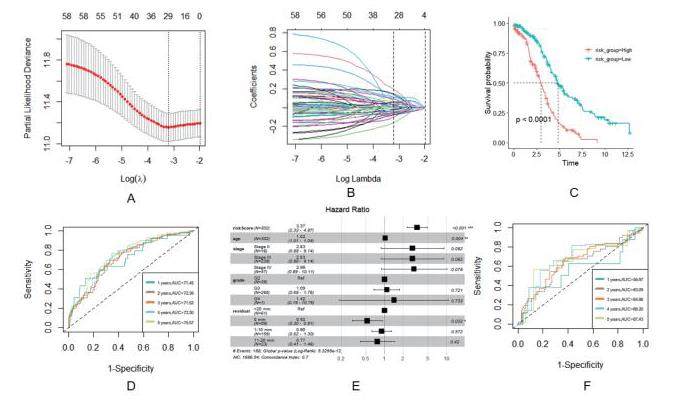
对TCGA数据集患者采用LASSO Cox回归法确定λ的最优值,鉴定出30个关键基因(图3.A.B)。然后根据30个关键基因的表达建立转移相关预后模型,并将其定义为转移评分,将患者的转移评分以中位数为标准分为高分组与低分组,Kaplan-Meier生存曲线显示两组间存在差异显著(图3.C),高评分组的生存率低于低评分组,同时TimeRoc曲线表明转移评分对患者预后具有可较好的预测(图3.D)。转移评分与其他临床因素进行单因素及多因素分析表示转移评分、年龄及残余病灶大小可作为卵巢癌的独立预后因素(图3.E)在GSE9891数据集中,转移评分也可对患者预后进行可较好的评价(图3.F)。
预后模型的建立与验证。(A)通过交叉验证选择LASSO 回归的最优参数λ(B)根据最优参数(λ)建立的含30个基因的预后模型。(C)高低转移评分组的生存分析。(D)预后模型对1至5年生存率的预测。(E)转移评分与其他临床因素的森林。(F)预后模型对GSE9891数据集中患者1至5年生存率的预测。
3.讨论
卵巢癌是女性生殖道普遍存在的恶性肿瘤,大多数患者诊断是已为晚期诊断,因此5年生存率极低。目前,OC的推荐治疗方法是手术联合铂类药物、紫杉醇化疗,大多数患者最初在化疗后会出现临床完全缓解,约有30%~40%的少数患者在接受治疗后仍会出现不完全缓解或疾病进展,但大多数患者最终会复发并伴有远处转移[14]。
对转移相关基因进行GO、KEGG分析,提示转移多与细胞基质相关。对TCGA样本就影响预后的转移基因进行无监督聚类分析,可分为两亚型。对两亚型进行通路分析,一亚组主要与DNA修复相关,抑制癌变,具有较好预后,另一亚型则多富集于细胞基质通路,预后较差。考虑转移相关基因对OC异质性及相应临床结局的影响,构建了包含30个基因的模型,以量化转移评分。评分低的患者比转移评分高的患者生存率更高,作为卵巢癌患者的独立预后生物标志物,多个数据集验证后表明对卵巢癌患者的预后具有一定的预测能力。
该研究是基于公开数据的回顾性研究,生物标志物的临床应用需要更多样本的数据支持。
参考文献
[1] Siegel, R.L., Miller, K.D. and Jemal, A. ‘Cancer statistics, 2018’, CA: A Cancer Journal for Clinicians, 68(1), pp. 7–30. doi:10.3322/caac.21442.
[2] Sung, H. et al. ‘Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries’, CA: A Cancer Journal for Clinicians, 71(3), pp. 209–249. doi:10.3322/caac.21660.
[3] Smith, L.H. et al. ‘Ovarian cancer: Can we make the clinical diagnosis earlier?’, Cancer, 104(7), pp. 1398–1407. doi:10.1002/cncr.21310.
[4] Ritchie ME, Phipson B, Wu D. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007.
[5] Wilkerson, M.D. and Hayes, D.N.‘ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking’, Bioinformatics, 26(12), pp. 1572–1573. doi:10.1093/bioinformatics/btq170.
[6] Hänzelmann, S., Castelo, R. and Guinney, J. ‘GSVA: Gene set variation analysis for microarray and RNA-Seq Data’, BMC Bioinformatics, 14(1). doi:10.1186/1471-2105-14-7.
[7 ]Gao, J., Kwan, P.W. and Shi, D. ‘Sparse kernel learning with LASSO and bayesian inference algorithm’, Neural Networks, 23(2), pp. 257–264. doi:10.1016/j.neunet.2009.07.001.
[8] Bookman, M. A., Okamoto, A., Stuart, G. Harmonising clinical trials within the Gynecologic Cancer InterGroup: consensus and unmet needs from the Fifth Ovarian Cancer Consensus Conference. Annals of oncology : official journal of the European Society for Medical Oncology, 28(suppl_8), viii30–viii35.
作者简介:朱顺鹏,男,安徽滁州,苏州大学附属第一医院妇产科研究生在读,研究方向为妇科肿瘤

 京公网安备 11011302003690号
京公网安备 11011302003690号


