


- 收藏
- 加入书签

基于线粒体基因组的金花菌与近缘种条形码筛选
 打开文本图片集
打开文本图片集
摘要:测定并注释了金花菌JH1805线粒体全基因组序列,探讨线粒体基因组不同序列位点开发分子标记的可行性,对6个长度在600~1800 bp的线粒体基因cob、cox1、cox2、cox3、nad2、nad6开发分子标记的潜力进行评估,其中以cox1分辨率最高,但对少数物种如Aspergillus flavus、Aspergillus ustus的鉴别效果不佳。鉴于此,本研究开发了nad6+cox1组合序列分子标记,可更有效地区分一些单基因无法鉴别的物种,用其所构建的系统树分枝置信度较高,在曲霉及青霉属物种的鉴别中适用性较好。
金花菌是黑茶发酵过程中的优势微生物,随着黑茶产业的崛起而为人们所熟知,目前,对金花菌的分类研究主要集中在其生理特性、发酵工艺、代谢产物等方面,而与金花菌及其近缘种鉴定分类相关的研究存在着一定局限:(1)菌株所表现的培养特征受环境因素影响,存在基因表达不一致的现象,使形态学鉴定的结果不稳定;(2)目前所用的分子标记序列(如ITS、18S rDNA等)相对保守,能提供的系统发育学聚类信息较少,尤其对于近缘程度较高的单位区分较为困难。此外,金花菌与它的2个近缘种——假灰绿曲霉(Aspergillus pseudoglaucus)、谢瓦氏曲霉(A. chevalieri)在分布环境、菌落特征、孢子形态上都有着较高的相似度,对其系统分类和研究造成了一定的困难,三者常在概念上被混淆[1-3],这对金花菌的菌种管理及应用是不利的。
与核基因组相比,线粒体DNA具有同源基因数量多、对突变压力敏感、进化速度快等特点[4-5],因此更适宜用于区分亲缘关系较近的物种和探究较短时间周期内发生的进化事件。此外,线粒体DNA的基因序列较多是中性进化分子,不同基因有着不同的进化速度及规律,通过适宜序列片段的筛选便可有效阐明物种间的进化关系[6],即使是长度较短的基因片段,也被证实能在物种鉴定中得到较为精确的结果[7],在第二代高通量测序技术的支撑下,线粒体全基因组测序工作变得高效便捷。鉴于此,研究拟通过对金花菌及其近缘种线粒体DNA的不同序列进行分析,以期筛选能够有效鉴别其种间单位的DNA条形码,旨在进一步明晰金花菌及其种属间的分子分类学问题,为促进茶叶发酵优势微生物种群的群体遗传学、进化和分类学等提供依据。
1试验方法
1.1 线粒体DNA提取、测序与注释
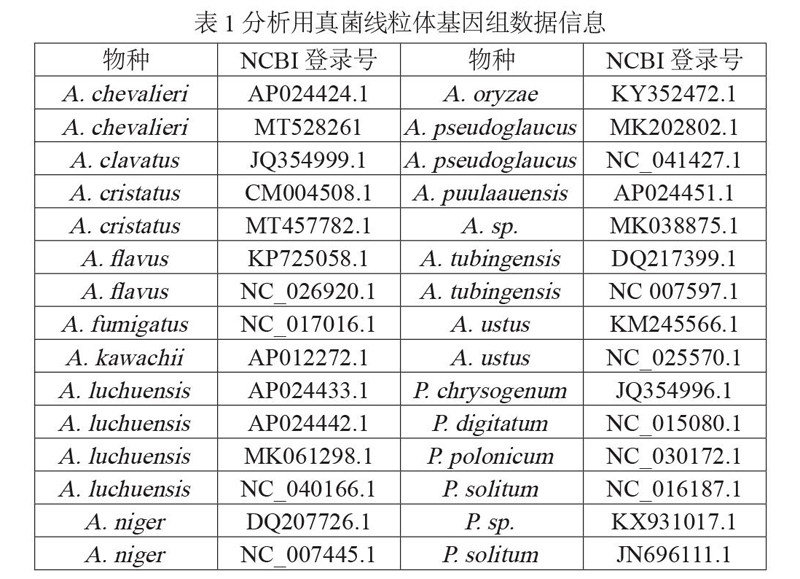
除自行测序的金花菌JH1805线粒体全基因组序列外,从NCBI核酸库下载近缘种真菌线粒体DNA序列,用于DNA条形码的筛选分析,相关真菌线粒体基因序列信息如表1所示。
1.2 分子标记候选序列的筛选
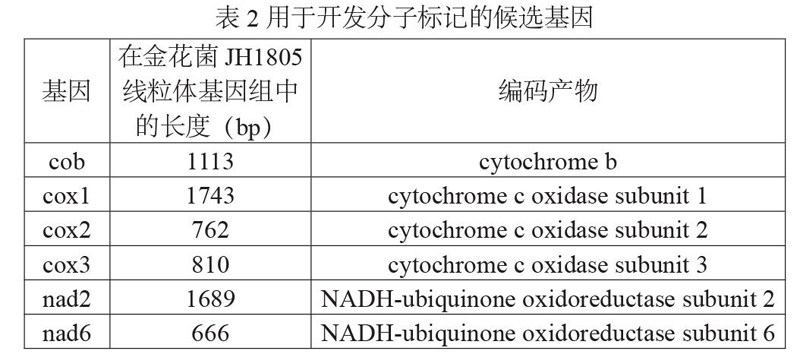
金花菌及其近缘种所共有的线粒体基因包括cob、nad1、nad4、atp8、atp6、nad6、cox3、cox1、atp9、nad3、cox2、nad4L、nad5、nad2、lrRNA、srRNA 16个序列,结合以往研究对真菌线粒体分子标记基因的筛选结果[8-9],并综合考虑PCR扩增及产物后续检测的需求,选取cob、cox1、cox2、cox3、nad2、nad6 6个长度在600~1800 bp的基因作为开发金花菌及近缘物种分子标记的候选序列(表2)。
1.3不同序列遗传距离及系统发育分析
采用FeatureExtract 1.2L从30个真菌线粒体基因组中提取cob、cox1、cox2、cox3、nad2、nad6基因序列信息[10],用Mega 5.0将各序列对齐后,使用双参数法计算不同真菌同一序列的遗传距离[11],并基于各序列信息,采用Maximum Likelihood法,构建系统发育树,自举检验值(boot-strap test)设置为1000。
2 结果与分析
2.1 不同序列种内、种间遗传距离
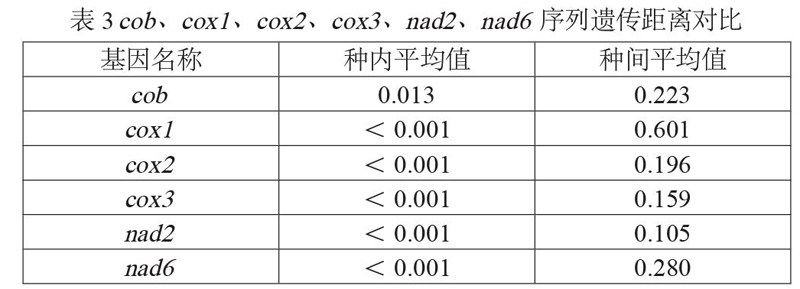
遗传距离是用于衡量物种间变异程度大小的标尺,分类阶元越高,遗传距离越大,可为传统分类学提供有效的依据[12]。统计30株真菌种内、种间平均遗传距离值,如表3所示。种内遗传距离除cob为0.013外,其余均小于0.001,满足Hebert等[13]所提出的同一物种种内遗传距离应小于0.02的条件;cob、cox1、cox2、cox3、nad2、nad6 6个基因序列的种间遗传距离平均值分别为0.223、0.601、0.196、0.159、0.105、0.280,均超过种内遗传距离平均值10倍以上,满足分子标记序列所需的种间遗传距离/种内遗传距离>10的要求[13],鉴于此,所选6个基因序列均符合开发分子标记的基本条件。此外,由进化速率cox1>nad6>cob>cox2>cox3>nad2可知,cox1与nad6具有更高的分子标记开发潜力。
2.2 基于cob、cox1、cox2、cox3、nad2、nad6序列的系统发育树
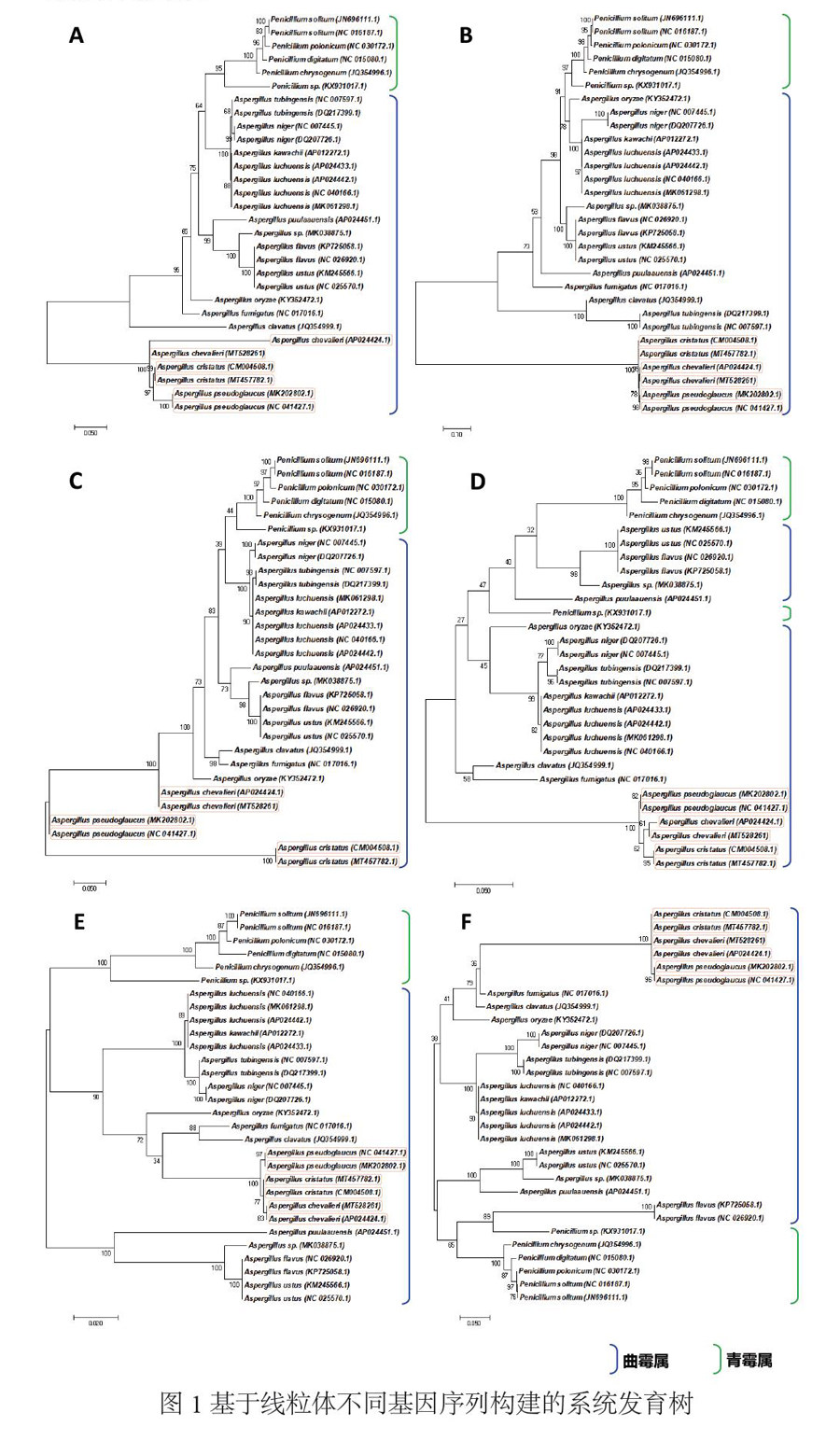
由于线粒体不同基因的进化速度存在差异,所构建的进化树拓扑结构也因此而不同。基于线粒体cob、cox1、cox2、cox3、nad2、nad6序列,采用Maximum Likelihood法构建的系统进化树如图1所示,6个线粒体基因作为分子标记均能一定程度区分Penicillium属、Talaromyces属和Aspergillus属物种之间的关系,使得不同单位基本聚集在有效的子分支上,且6个基因所构建的系统树拓扑结构大致相符,所揭示的物种系统发育关系相似度较高。
但是,利用单基因作为曲霉属及其近缘物种分子标记仍然存在着一些不足之处,主要包括:(1)除nad6外,其余序列均无法有效区分A. flavus与A. ustus;(2)cob序列构建的系统树在A. chevalieri与A. chevalieri的划分上存在异常,二者间遗传距离达到了0.182,超于种内距离平均值;(3)nad6序列所构建的系统树无法有效区分A. chevalieri与A. cristatus,二者被划分为同一小支。
此外,还有一些基因序列显示的物种间遗传距离过小,远低于物种间差异2%的遗传距离[13],从而无法在系统树上形成显著的分支,如:(1)cob序列构建的系统树无法较好的区分A. tubingensis、A. luchuensis与A. niger(遗传距离值0.003~0.004);(2)cox2构建的系统树无法有效区分A. luchuensis与A. tubingensis(遗传距离为0.004)。因此,还需要试验多基因序列的组合,来实现更为为准确的物种间区分。
2.3 基于拼接序列nad6+cox1的系统发育树
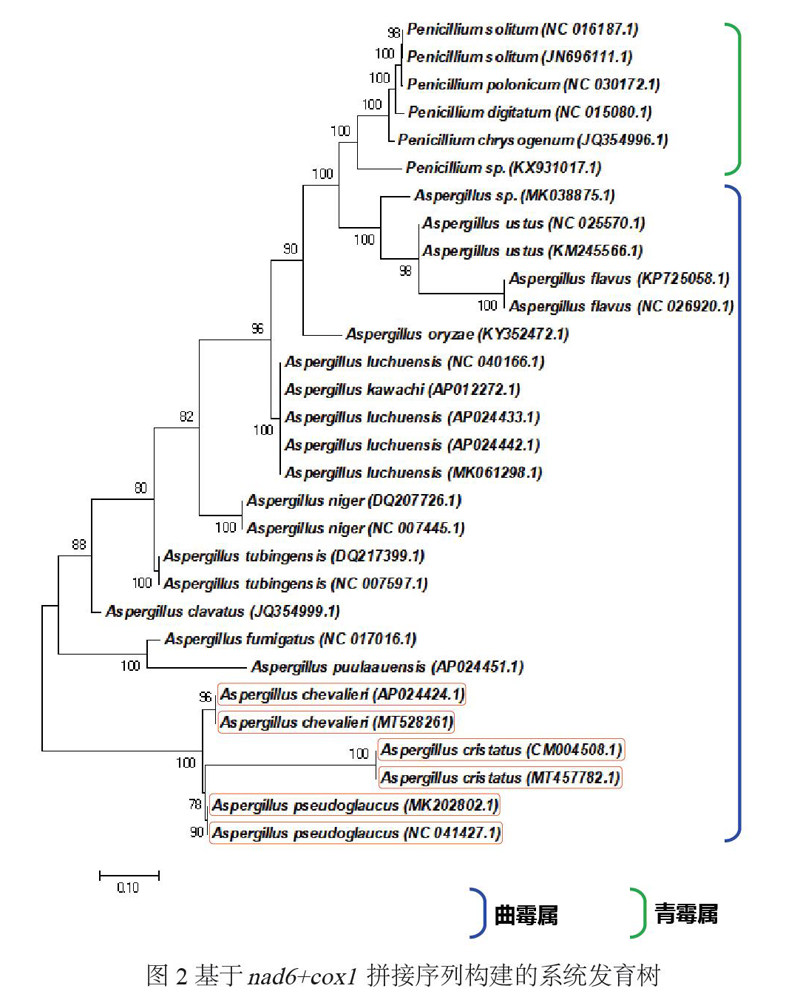
为了开发更准确的线粒体序列分子标记,根据单基因对物种鉴别的结果,进一步采用nad6+cox1拼接序列计算遗传距离并构建系统进化树,系统树如图2所示、遗传距离见附录9。由结果可知,nad6+cox1序列构建的系统树,其物种间关系分辨率较高,相同的种均被划分到了同一分支下,不同物种间可形成明显的条形码间隙,且自展分析的节点支持率较高。该拼接序列种间遗传距离为0.388,种内遗传距离<0.001,能较好的区分种内/种间单位,适宜用于曲霉及其近缘种属遗传差异的分析。此外,黑茶中的3种近缘微生物金花菌、谢瓦氏曲霉、假灰绿曲霉相互之间的遗传距离均超过了2%的种间距离值,可依据此组合序列条形码较好的区分开来。虽然A. kawachii与4株A. luchuensis在系统树上被聚在同一小支上,但根据Yamada等[14]的研究,二者实为同一真菌的不同名称。
图2 基于nad6+cox1拼接序列构建的系统发育树
3讨论
近年来,DNA条形码被广泛的应用于物种分类鉴定、群体遗传关系等方面的研究[15]。针对曲霉属物种不同环境下表型差异较大、鉴别困难的问题,本研究对线粒体DNA序列开发分子标记的可行性进行了探讨,筛选了cob、cox1、cox2、cox3、nad2、nad6 6个长度在600~1800 bp的基因作为候选序列,通过进化速率计算及系统发育分析,结果显示,6个单基因序列所构建的系统树拓扑结构大致相同,其中cox1的进化速度最快,种间平均遗传距离达到0.601,对除A. flavus、A. ustus之外的真菌有着较高的分辨率,并且序列长度较为适宜,是适用性较好的单基因序列分子标记,这与Damon[16]、Yang[17]对Pezizomycotina、Laetiporus sulphureus等真菌进行分子标记开发获得的结论一致。
针对一部分菌株无法被单基因序列标记的不足,研究探讨了组合序列nad6+cox1开发分子标记的可行性,结果表明,二种标记组合可有效区分所有供试菌株,用其所构建的系统树分枝置信度较高,能较好的反映曲霉属及其近缘单位进化关系。通过线粒体序列复合条形码技术,可实现对真菌丰富的物种及高级分类单元多样性的准确鉴别,符合时下真菌分类学快速、准确和标准化的要求[18],具有较高的应用潜力。
参考文献
[1] 孟雁南. 陕西茯砖茶优势菌群及其功能性研究[D]. 陕西科技大学, 2019.
[2] 王磊, 谭国慧, 潘清灵, 等. 黑茶砖茶中两种产生“金花”的曲霉菌[J]. 菌物学报, 2015, 34(2): 186-195.
[3] 龙章德,陈皓睿,刘鸿, 等.来自六堡茶的3株散囊菌属真菌对烟草的发酵效果分析[J].南方农业学报,2018,49(10):2055-2061.
[4] Alexeyev M, Shokolenko I, Wilson G, et al. The maintenance of mitochondrial DNA integrity—critical analysis and update[J]. Cold Spring Harbor perspectives in biology, 2013, 5(5): a012641.
[5] Basse C W. Mitochondrial inheritance in fungi[J]. Current Opinion in Microbiology, 2010, 13(6): 712-719.
[6] Sulaiman Z H, Hui T H, Lim K K P, et al. Mitochondrial DNA sequence analyses in Bornean sucker fishes (Balitoridae: Teleostei: Gastromyzontinae)[J]. Integrative zoology, 2006, 1(1): 12-14.
[7] Min X J, Hickey D A. Assessing the effect of varying sequence length on DNA barcoding of fungi[J]. Molecular Ecology Notes, 2010, 7(3): 365-373.
[8] 林惠娇, 蒋湘, 王新国, 等. DNA条形码技术及其在真菌研究中的应用[J]. 植物检疫, 2013, 27(2): 11-18.
[9] Vialle A, Feau N, Allaire M, et al. Evaluation of mitochondrial genes as DNA barcode for Basidiomycota[J]. Molecular Ecology Resources, 2010, 9(s1): 99-113.
[10] Wernersson R. FeatureExtract—extraction of sequence annotation made easy[J]. Nucleic Acids Research, 2005, 33(suppl 2): W567-W569.
[11] Kimura, Motoo. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences[J]. Journal of molecular evolution, 1980, 16(2): 111-120.
[12] Amrita, Srivathsan, Rudolf, et al. On the inappropriate use of Kimura-2-parameter (K2P) divergences in the DNA-barcoding literature[J]. Cladistics, 2012, 28: 190-194.
[13] Hebert P D N, Cywinska A, Ball S L, et al. Biological identifications through DNA barcodes[J]. Proceedings of the Royal Society B: Biological Sciences, 2003, 270(1512): 313-321.
[14] Yamada O, Takara R, Hamada R, et al. Molecular biological researches of Kuro-Koji molds, their classification and safety[J]. Journal of Bioscience & Bioengineering, 2011, 112(3): 233-237.
[15] 曾昭清,赵鹏,罗晶, 等.从真菌全基因组中筛选丛赤壳科的DNA条形码[J].中国科学:生命科学,2012,42(1):55-63.
[16] Damon C, Gérard Barroso, Cyril Férandon, et al. Performance of the COX1 gene as a marker for the study of metabolically active Pezizomycotina and Agaricomycetes fungal communities from the analysis of soil RNA[J]. FEMS Microbiology Ecology, 2010,74(3):693-705.
[17] Li Q, Yang M, Chen C, et al. Characterization and phylogenetic analysis of the complete mitochondrial genome of the medicinal fungus Laetiporus sulphureus[J]. Scientific reports, 2018, 8(1): 1-12.
[18] 张宇,郭良栋.真菌DNA条形码研究进展[J].菌物学报,2012,31(6):809-820.
作者简介:易諝怀(2001—),男,本科生,高分子材料与工程专业
基金项目:湖南省自然科学基金项目(2021JJ40021),湖南城市学院大学生创新创业训练计划项目(S202211527064),湖南省教育厅科学研究项目(20B112),湖南省大学生创新创业训练计划项目(S202111527051)

 京公网安备 11011302003690号
京公网安备 11011302003690号


